Research Summary
Research in the CRUNCH LAB lives at the fertile intersection of computational science, nanotechnology, and clean energy — three of the most interesting and important areas of current science and engineering. In particular, with NSF funding (NSF-1512442) we model the formation of biofuels in nanoporous zeolite catalysts, and with DOE funding (DE-SC0019170) we simulate the self-assembly and crystallization of zeolite nanopores. We also study structures and dynamics of hydrogen-bonding networks of interest to next-generation fuel cells. In what follows, we describe the grand-challenge questions and promising new methods being investigated in these projects. In particular, we are pursuing research in the following areas:
- Making Clean Biofuels in Zeolite Catalysts – From “Grass to Gas”
- The Mystery of Zeolite Nanopore Self-Assembly – Seeing “Invisible” Patterns
- Proton Transfer in Fuel Cells – In Search of a “Liquid-like Solid”
Making Clean Biofuels in Zeolite Catalysts – From “Grass to Gas”
 |
Biofuels are liquid transportation fuels produced from plant biomass. Such clean fuels offer the
promise of carbon neutral transportation energy because the carbon dioxide soaked up by
growing fuel crops can balance the carbon dioxide generated during fuel combustion. Producing
cheap biofuels relies on selective catalyst systems for converting biomass-derived feedstocks
into high octane fuels such as gasoline, diesel, and jet fuels. Zeolites are the most heavily used
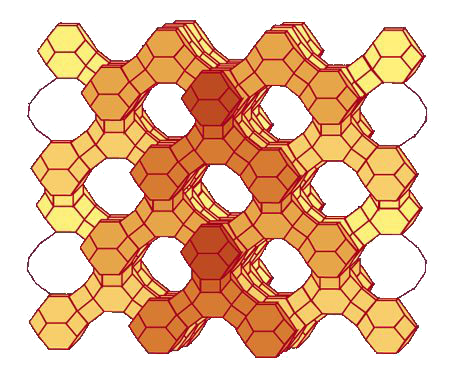
catalysts on planet earth, and show promise for making an array of biofuels(see Fig. 1). However, at present,
too much of the biomass carbon is lost to carbonaceous deposits known collectively as “coke”
during zeolite processing.
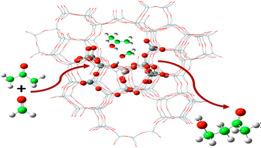
Computational research in the CRUNCH LAB is focused on discovering the chemical pathways that
convert biofuel precursors into coke species, and how these pathways can be turned off. We
work with NSF support and in collaboration with the spectroscopy group of F. Jentoft in
Chemical Engineering at UMass Amherst. We have developed novel cluster methods for
speeding up quantum calculations [1,2], and are presently applying computational spectroscopy
and accelerated ab initio dynamics methods to identify key coke-forming mechanisms. Our goals
are to find and close the bridge that connects biofuels to coke, to make biofuel production more
efficient and thus more abundant in global energy markets.
- [1]Angela Migues, Adina Muscat, Scott M. Auerbach, Woody Sherman, and S. Vaitheeswaran, “On the Rational Design of Zeolite Clusters,” ACS Catalysis 5, 2859-2865 (2015). [PDF]
- [2] Angela N. Migues,† Qinfang Sun,† S. Vaitheeswaran, Woody Sherman, and Scott M. Auerbach, “On the Rational Design of Zeolite Clusters for Converging Reaction Barriers: Quantum Study of Aldol Kinetics Confined in HZSM-5,” J. Phys. Chem. C 122,23230-23241 (2018). (†Co-first authors) [PDF]
The Mystery of Zeolite Nanopore Self-Assembly – Seeing “Invisible” Patterns
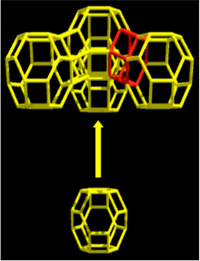 |
One of the great mysteries in nanotechnology is how zeolites form in solution(see, e.g., Fig. 2). Given that “nature abhors a vacuum,” it is natural to wonder how open nanopores can form? Our ignorance about the mechanism of zeolite formation severely hampers our ability to synthesize new, tailor-made zeolites for advanced applications such as biofuel production (see project above) and carbon dioxide capture. We do know that zeolite formation requires a “structure directing agent” (SDA) in solution, but the exact role of the SDA is poorly known given that there is rarely a one-to-one mapping between a particular SDA and an eventual zeolite framework structure. We hypothesize that SDAs (and also heteroatoms such as Al, Ge and Zn that replace Si in the framework) can influence the rings present in the pre-crystalline synthesis gel. With DOE funding, and in close collaboration with W. Fan (UMass Chemical Engineering), we are testing this hypothesis through an experimental and modeling research program integrated with Raman and total scattering spectroscopies as probes of structure of the amorphous synthesis gel. Our hierarchical modeling effort involves the unique combination of quantum chemistry for predictive accuracy, molecular dynamics of solution interactions, and our own reaction ensemble Monte Carlo approach for simulating the assembly of silica-SDA nanoparticles, synthesis gels, and zeolite crystals [3]. We aim to reveal new design principles for zeolite synthesis, to usher in a new era in zeolite science of tailor-made materials by design.
Recent publications:
- [3] Scott M. Auerbach, Wei Fan, and Peter A. Monson, “Modeling the Assembly of Nanoporous Silica Materials,” (Invited by) International Reviews in Physical Chemistry 34, 35-70 (2015). [PDF]
Proton Transfer in Fuel Cells – In Search of a “Liquid-like Solid”
 |
Hydrogen fuel cells represent a promising, light-weigh/bt technology for efficient conversion of fuels to electricity. In the USA in 2017, the efficiency of generating electricity was 33.6%, meaning that 66.4% of the fuel energy (in natural gas and coal) was wasted. The magnitude of this wasted energy equals more than 31 times all the solar electricity generated in the USA during 2017!! Hydrogen fuel cells provide a promising technology for sharply reducing this wasted energy, by converting the chemical energy of H2 into electrical energy without burning, and with efficient catalysts. Essential to the hydrogen fuel cell is a membrane that separates H2 and O2, and that rapidly shuttles protons from anode to cathode. Liquid phosphoric acid holds the world record for proton conductivity because of its facile fluctuations and polyprotic connectivity. Herein lies the fundamental problem: we seek a mechanically-stable, solid membrane that also exhibits high proton conductivity, requiring liquid-like fluctuations at the molecular scale (like phosphoric acid) — i.e., a liquid-like solid.
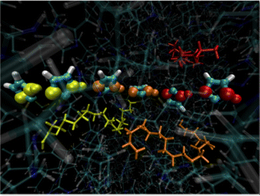
Towards this end, we have modeled hydrogen-bonding network structures and dynamics of tethered amphiprotic molecules via molecular dynamics simulations(see Fig. 3) [4]. In particular, we have analyzed the trade-off between well-aligned hydrogen-bonding systems, and rapidly rotating functional groups, to provide new design criteria for advanced proton exchange membranes. This work is challenging because of the need for both chemical accuracy and statistical sampling of oligomer configurations. Our work will yield new design ideas for advanced proton exchange membranes in next-generation fuel cells.
Recent publications:
- [4] Qinfang Sun, Jacob A. Harvey, Katharine V. Greco, and Scott M. Auerbach, “Molecular Simulations of Hydrogen-Bond Cluster Size and Reorientation Dynamics in Liquid and Glassy Azole Systems,” J. Phys. Chem. B 120, 10411-10419 (2016). [PDF]
Updated November 2018