B.S. 1988, Georgetown University, Summa cum Laude; Ph.D. 1993, UC Berkeley; NSF Postdoctoral Fellow, 1994-1995, UC Santa Barbara Materials Research Laboratory
Physical Chemistry; Theoretical studies of molecular adsorption, diffusion and reaction in inorganic and biological nanopores; quantum, classical and statistical mechanics of solid-fluid interfaces.
Tel: (413) 545-1240; Internet: auerbach@chem.umass.edu

Principal Research Interests
My research group is focused on modeling catalysis and transport in nanoporous molecular sieves, such as zeolites and other aluminosilicates. Although these materials lie at the heart of many industrial applications, such as petroleum refinement and air separation, the microscopic events that underlie their application are poorly known. Several fundamental questions arise: how do molecules react in zeolites? How do they diffuse through? Which process is faster? Can modifying a molecuar sieve alter the balance between diffusion and reactivity? The theoretical techniques developed in my group are beginning to answer such questions, helping to suggest new materials with interesting properties and advanced performance.
Our theoretical approach begins by calculating host-guest interactions with both empirical force field and quantum mechanical methods. We then compute hopping and reaction rate constants for molecules adsorbed in zeolites by applying classical and quantum rate theories. Finally, we model the intricate balance between diffusion and reaction with the kinetic Monte Carlo technique, which randomly selects processes with frequencies determined by the input rate constants. In the end, we learn which underlying rate process most controls zeolite catalysis.
We have developed a new force field for aromatics in zeolites, and have applied it to modeling benzene diffusion in the zeolites Na-X, Ca-X, Na-Y, and siliceous Y. The S(II)->W process (pictured below) controls diffusion in these materials. We have also developed an analytical theory for the concentration dependence of benzene diffusion in faujasite, which demonstrates excellent qualitative agreement with pulsed field gradient NMR data. We are performing quantum cluster calculations to study benzene-proton interactions in the acidic zeolite H-Y, to model benzene and proton mobilities. Calculations are also underway to study butene isomerization dynamics in the zeolite ferrierite.
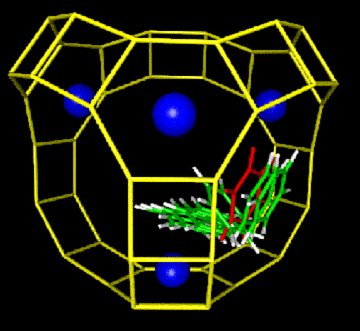
Microscopic hopping mechanism (with energy profile below) controlling macroscopic diffusion of benzene in faujasite, the major zeolite catalyst used in petroleum refinement.
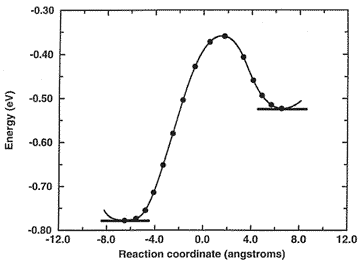 Energy Profile
Energy Profile